뉴스 검색결과 10,000건 이상
- 엔허투, 세계 최초 암종 불문 치료제 '성큼'[제약·바이오 해외토픽]
- [이데일리 신민준 기자] 항체-약물접합체(ADC) 항암제인 엔허투가 세계 최초 암종 불문 치료제에 성큼 다가서고 있다. 엔허투는 미국 식품의약국(FDA)으로부터 모든 고형암에 대한 치료제로 승인을 받았다. 엔허투. (사진=다이이찌산쿄)14일 외신과 한국바이오협회에 따르면 미국 식품의약국은 최근 엔허투를 상피세포증식인자수용체 2형(HER2) 양성 고형암 치료제로 가속 승인했다. 엔허투는 다이이찌산쿄와 아스트라제네카가 공동 개발한 항체-약물접합체 항암제다. 엔허투는 허셉틴으로 알려진 기존의 HER2 타깃 항체 트라스투주맙 성분에 다이이찌산쿄가 개발한 세포독성물질(페이로드) 데룩스테칸이 결합해 만들어졌다. 트라스투주맙이 HER2 암세포를 찾아내 페이로드가 암세포만 공격한다. 미국 식품의약국은 객관적 반응률과 반응기간을 근거로 엔허투의 가속 승인을 결정했다. 향후 엔허투는 확증 임상을 통해 정식 허가 여부가 결정된다. 엔허투는 3건의 다기관 임상2상 연구에서 유효성이 확인됐다. 엔허투는 이전에 치료받은 절제 불가능 또는 전이성 HER2 양성 고형 종양이 있는 성인 환자 192명을 대상으로 세 가지 다기관(DESTINY-Pantumor02, DESTINY-Lung01, DESTINY-CRC02) 임상2상 시험 중 하나에 등록해 유효성을 확인했다. DESTINY-PanTumor02 임상 2상에서 △담관암 △방광암 △자궁경부암 △자궁내막암 △난소암 △췌장암 또는 기타 각종 종양을 엔허투로 치료받은 HER2 양성 고형암 환자들은 51.4%의 객관적 반응률(ORR, 사전에 정의된 최소한의 기간 동안 일정량 이상의 종양 감소를 보인 환자의 비율)과 19.4개월의 평균 반응기간(DOR)을 보였다. DESTINY-Lung01에서의 ORR은 52.9%, DOR 6.9개월이었고, DESTINY-CRC02에서의 ORR은 46.9%, DOR은 5.5개월 이었다.이 같은 암종 확대가 가능한 건 엔허투가 HER2 단백질을 타깃으로 하기 때문이다. HER2는 세포 내부로 신호를 보내 암 성장을 자극한다. HER2는 발현 수준이 높을수록 암의 재발·전이가 잦아 위험도가 높은 것으로 전해진다. HER2를 타깃하면 암의 진전을 억제할 수 있다. HER2 단백질의 과발현 현상은 주로 유방암, 위암, 폐암, 대장암에서 나타난다. 엔허투의 지난해 매출은 25억7000만달러(약 3조6000억원)를 기록했다. 이는 전년(12억5000만달러·약 1조7300억원)과 비교해 두배 이상 증가했다.
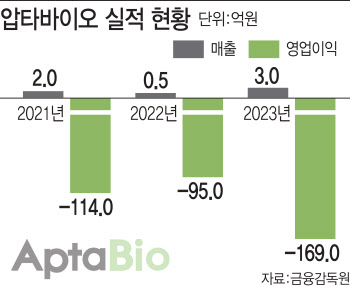
- 압타바이오, 올해 ‘APX-115’ 美 임상 2상 결과 주목
- [이데일리 김새미 기자] 압타바이오(293780)가 올해에는 글로벌 기술이전 성과를 통해 기업가치를 입증할지 주목된다. 핵심 파이프라인 ‘APX-115’(아이수지낙시브)의 조영제 유발 급성신장손상(CI-AKI) 미국 임상 2상 결과가 중요한 지표가 될 것으로 예상된다.◇연매출 30억원 요건이 걸림돌…자금은 넉넉10일 바이오업계에 따르면 압타아비오는 올해 연매출 30억원을 달성하지 않으면 관리종목으로 지정될 수 있다. 압타바이오는 2019년 6월 기술특례상장을 통해 코스닥 시장에 입성했기 때문에 기술특례로 매출 요건이 면제되는 기간은 올해 6월까지다. 관리종목으로 지정되면 상장폐지 실질심사 대상이 된다.그 외에 재무적인 리스크는 당분간 없을 것으로 전망된다. 지난해 389억원 규모의 전환사채(CB) 발행과 111억원 규모의 제3자배정 유상증자를 통해 총 500억원의 자금을 조달했다. 압타바이오의 3년간 판매관리비 평균치가 130억원 정도인 것을 고려하면 2년은 여유가 있다. 여기에 현금성자산(단기금융상품 포함) 440억원까지 더하면 940억원의 여유 자금이 있기 때문에 7년 이상 운영에 문제 없을 것으로 예상된다.[그래픽=이데일리 김일환 기자]단 연매출 30억원 요건은 신경써야 할 시점이다. 압타바이오는 최근 7년간 매출 30억원을 넘긴 적이 없다. 압타바이오의 매출은 2017년 3억원→2018년 15억원→2019년 10억원→2020년 3억원→2021년 2억원→2022년 4억원→2023년 3억원으로 상당히 저조하다.이 때문에 압타바이오도 특단의 조치를 취했다. 기존에 압타바이오의 사업 전략은 플랫폼 기술을 통한 공동연구 또는 기술이전을 통한 매출 발생에 중점을 뒀지만 지난해 11월 신사업본부를 신설, 안전장치를 마련한 것이다. 압타바이오는 건강기능식품 주문자상표부착생산(OEM) 사업과 펫케어 사업을 새로 추진하면서 연매출 30억원을 넘길 계획이다. 실제로 지난해 2개월이 안 되는 기간에 해당 사업으로 3억원의 매출이 발생했다는 게 회사 측의 설명이다.◇시장의 의구심, 글로벌 기술수출로 해소할 때중장기적으로는 글로벌 기술이전을 통한 성과가 본격화돼야 기업가치를 입증할 수 있을 것으로 보인다. 압타바이오의 시가총액은 1383억원에 불과하다.이처럼 시총이 낮은 이유는 아직 실질적인 글로벌 기술이전 성과가 없다는 시장의 평가 때문으로 추정된다. 압타바이오는 2016년 미국 호프바이오사이언스(Hope Bioscience)에 췌장암 치료제 ‘Apta-12’를 기술이전한 적이 있지만 2022년 1월 권리반환됐다. 기술이전 계약 규모도 비공개했기 때문에 어느 정도 가치를 인정받았는지 확인하기도 어렵다. 현재로서는 글로벌 기술이전 실적이 없는 셈이다.압타바이오가 “글로벌 기술력을 인정받아 조기에 3건의 기술이전 성과를 이뤘다”고 하지만 호프바이오사언스를 제외한 나머지 2건은 국내 제약사 삼진제약(005500)과 체결한 기술이전 계약이다. 그 마저도 삼진제약에 신약후보물질들을 기술이전한 시기가 2016년, 2018년으로 5년 이상 됐는데도 아직 임상 1상 단계라 협업이 지지부진한 것 아니냐는 우려가 나온다.2016년 삼진제약에 기술이전된 혈액암 치료제 ‘Apta-16’(SJP1604) 임상 1상은 지난해 2월 종료됐어야 하지만 아직까지도 진행 중이다. 2018년 삼진제약에 넘긴 황반변성 치료제 ‘APX-1004F’(SJP1803/1804)도 2021년 11월 임상 1상에 진입, 2022년 11월에는 임상을 마칠 것으로 예상됐으나 아직 임상 1상 단계에 머물러 있다. 우려와 달리 양사 협업이 아예 중단된 것은 아니라는 게 업계 관계자의 전언이다.◇‘APX-115’ 美 임상 2상 결과가 중요한 이유그럼에도 압타바이오는 올해 중요한 임상 결과 발표를 앞두고 있는 만큼, 기술이전 성과가 임박했다고 보고 있다. 압타바이오는 연내 핵심 파이프라인 ‘APX-115’(아이수지낙시브)의 조영제 유발 급성신장손상(CI-AKI) 미국 식품의약국(FDA) 임상 2상 결과를 발표할 예정이다. 해당 임상 2상은 올해 상반기 종료를 목표로 했지만 의사 파업으로 인해 다소 지연되는 분위기다.회사에 따르면 APX-115의 CI-AKI 임상 2상을 통해 약효를 입증하면 다른 파이프라인의 가치도 동반 상승하는 효과를 얻을 수 있다. APX-115는 압타바이오의 NADPH 산화효소(NADPH oxidase, 이하 NOX) 고효능 스크리닝 기술을 기반으로 개발한 신약후보물질로, 인체내 활성화 산소 조절이 실패할 경우 발생하는 질환을 근본적으로 치료하는 것을 목표로 하기 때문이다. 특히 해당 임상이 성공하면 올해 진입할 APX-115의 당뇨병성신증 치료제 임상 2b상도 탄력을 받을 전망이다.최근 압타바이오는 신규 파이프라인 ‘AB-19’ 연구개발에도 힘을 쏟고 있다. AB-19는 황반변성 치료제 및 면역항암제로 개발할 계획이며, 모두 올해 하반기 미국 임상 1상에 진입하는 것을 목표로 하고 있다. AB-19는 주사제가 아닌 점안제 형태로 개발되는 만큼, 글로벌 황반변성 시장에서 충분한 경쟁력이 있을 것으로 보고 있다. 2019년 연구를 시작한 AB-19는 현재 전임상 단계에 있다.압타바이오 파이프라인 현황 (자료=압타바이오)아울러 압타바이오는 이미 다양한 기업들과 활발하게 파트너링 논의를 하고 있는 만큼, 글로벌 기술이전 성과가 곧 도출될 것으로 보고 있다. 압타바이오 측은 “이미 라이선스아웃 대상 기술과 파이프라인에 대해 30여 개 기업과 비밀유지계약(CDA)를 체결하고 여러 차례 파트너링 미팅을 진행했다”며 “NOX 저해제 관련 파이프라인 대부분은 2021년 임상 2a상이 완료됨에 따라 2022년 이후부터 본격적인 협의가 진행되고 있다”고 강조했다.다만 CDA 체결이 실제 기술이전 계약 성사로 이어지기까지는 물질이전(MTA) 계약, 텀싯 작성, 실사 등 많은 과정을 거쳐야 한다. CDA 체결은 기술이전 협의 초기 단계이기 때문에 실제 기술이전까지 이어질 가능성은 글로벌 평균 통계상 1.9% 정도다.

- ADC 신화 써내려가는 다이이찌산쿄, 입지 좁아지는 레고켐바이오
- [이데일리 김진호 기자] 일본 다이이찌산쿄와 영국 아스트라제네카(AZ)가 공동으로 항체약물접합체(ADC) 신약의 글로벌 상용화에 박차를 가하고 있다. Trop2를 차용한 ‘다토포타맙 데룩스테칸’으로 다시 한 번 후발주자를 멀찍이 따돌리겠다는 구상이다. 회사는 미국을 기점으로 각국에서 동시다발적으로 이 약물을 허가 심사대에 올려 놓겠다는 전략이다.다이이찌산쿄가 엔허투에 성공에 이은 또다른 ADC 신화를 써내려갈지 관심을 모은다. 회사는 최근 미국 머크(MSD)와 220억 달러 규모 계약을 체결하며 새로운 공동전선도 구축했다. 국내 레고켐바이오(141080)(새사명은 리가켐바이오)가 ADC 각 고형암 분야 2~3순위권 후발 개발주자로 이름을 올리고 있지만, 빅파마의 다각적인 확장 공세에 밀려 그 성장성이 위축될 수 있다는 우려가 나온다.일본 다이이찌산쿄가 영국 아스트라제네카와 유전자 변형 Trop2 타깃 ADC 후보물질에 대한 각국 허가절차에 돌입하는 동시에 미국 머크와의 새로운 연합전선으로 시장 장악을 시도하고 있다.(제공=각 사)8일 제약바이오 업계에 따르면 다이이찌산쿄와 AZ가 공동개발한 차세대 ADC인 다토포타맙 데룩스테칸이 미국에서 일부 유방암과 폐암 환자 대상 적응증 획득을 위한 최종 관문에 연이어 진입했다.지난 2일(현지시간) 미국 식품의약국(FDA)은 다토포타맙 데룩스테칸에 대해 호르몬 수용체(HR) 양성 및 상피세포성장인자수용체(HER)2 음성 절제불가성 전이성 유방암 적응증에 대한 다토포타맙 데룩스테칸의 허가 심가 건을 수락했다. 앞선 지난 2월 FDA는 비편평 조직유형을 가진 비소세포폐암 적응증에 대한 다토포타맙 데룩스테칸 허가 심사에도 착수했다. 늦어도 내년 1분기까지 해당 약물이 미국에서 2종의 적응증을 획득할 잠재력을 가지게 된 셈이다. HR 양성 HER2 음성 유방암 적응증은 다이찌산쿄의 전작인 엔허투(성분명 트라스투주맙 데룩스테칸)가 확보하지 못한 적응증이다. 엔허투는 미국과 EU 등에서 HER2 양성 유방암 및 위암과 HER2 저발현 유방암, HER2 변이 비소세포폐암 등 4종의 적응증을 보유하고 있다. 이에 다이이찌산쿄는 엔허투에 쓴 트라스투주맙 대신 유전자 변형 Trop2 타깃 항체인 다토포타맙을 적용한 ADC를 발굴했다. HER2 음성 유방암 환자는 전체의 75~80%로 알려졌으며, 이들에게서 폭넓게 발현하는 단백질이 Trop2다. 항체와 접합체, 톡신(페이로드) 등 ADC의 주요 구성요소 중 1가지를 변경한 다토포타맙 데룩스테칸으로 엔허투가 누리지 못한 시장까지 진출하려는 것이다.정보분석 기업 클래리베이트가 지난 1월 연례보고서에서 5년 내 10억 달러 이상 매출을 기록할 약물로 다토포타맙 데룩스테칸을 주목했다. 이 약물이 유방암과 비소세포폐암 적응증을 획득해 2029년경 27억 달러(한화 3조6000억원)이상 매출을 달성할 것으로 전망한 것이다. 이런 예상이 다소 낙관적이라는 의견도 나온다. ADC 개발 업계 관계자는 “HER2 음성을 포함한 삼중음성유방암으로 개발된 약물 ‘트로델비’ 역시 Trop2 항체다”며 “트로델비는 비소세포폐암 관련 적응증 확장 임상에서 유의미한 생존기간(OS)을 확보하지 못했다”고 꼬집었다. 실제로 지난 1월 미국 길리어드 사이언스는 자사 ADC ‘트로델비’(성분명 사시투주맙 고비테칸)의 폐암 적응증 확장 임상이 실패했다고 밝힌 바 있다.그는 이어 “다이이찌산쿄의 유전자 변형 항체인 다토포타맙이 유방암 적응증은 무리없이 넘을 수 있지만, 폐암 적응증을 획득할지는 지켜봐야 한다”고 꼬집었다. 이어 “엔허투 개발사라는 후광에 힘입어, 여러모로 성공할 것이란 전제가 포함된 매출 전망치인 것 같다”고 말했다. 그럼에도 Trop2 타깃 ADC 중 트로델비에 이어 두 번째로 시장에 등장할 약물로 다토포타맙 데룩스테칸이 유력한 물질임에는 변함이 없는 상황이다. ◇뒤쫓는 레고켐에 닿지 않는 다이이찌산쿄다이이찌산쿄가 걸어간 길을 뒤따르고 있는 기업이 레고켐바이오다.레고켐바이오는 미국에서 Trop2-ADC 후보물질 ‘LCB84’의 고형암 대상 임상 1/2상을 자체적으로 진행하고 있다. 이외에도 회사가 확보한 트라스투주맙 기반 ADC인 ‘LCB14’는 임상 초기 단계에서 엔허투를 능가하는 효능을 보였다. LCB14는 현재 글로벌 임상(HER2 유방암 대상 중국 임상 3상 및 호주 임상 1상 등)에 올라 있는 상태다. 하지만 그 성과를 제대로 보여줄 새도 없이 빅파마의 폭풍공세가 이어지고 있다. 다이이찌산쿄는 물론 화이자에 인수된 ‘시젠’, ‘티브닥’(티소투맙 베도틴) 개발에 성공한 덴마크 젠맙 등 ADC 선도 기업들이 빅파마와 연합전선을 구축한 지 오래다. 이를 통해 앞서 언급한 다토포타맙 데룩스테칸과 같은 신규 ADC 개발도 속속 가시권에 들어오고 있다. 최근 MSD는 면역항암제인 차세대 성장 동력으로 ADC를 꼽았다. MSD는 다이이찌산쿄가 발굴한 3종의 ADC 신약 후보물질의 글로벌 개발 및 상업화 권리를 확보하기 위해 총 220억 달러(약 30조원) 규모의 계약을 체결했다.바이오 업계 한 임원은 “AZ가 엔허투의 개발 권리를 가져가서 빠르게 글로벌 상업화에 성공했다”며 “이번에는 항암제 제왕이라 불리는 ‘키트루다’를 보유한 MSD와 손잡으면서 그 가능성이 재주목받고 있다”고 말했다. 이어 “젠맙 역시 티브닥을 공동개발한 시젠이 화이자에 흡수된 만큼 이들과 긴밀히 연결된 상태다, 그 외에도 젠맙은 미국 애브비 등과도 다양한 ADC를 개발하고 있다”고 말했다.일각에서는 국내 바이오텍의 성장을 위해 세계적인 기업에게 보다 적극적으로 자사의 연구 성과나 후보물질 정보를 알려야 한다는 이야기가 나온다. 묵현상 퍼스트바이오이사회 의장(전 국가신약개발재단 단장)은 “바이오텍이 초기 연구성과를 해외 제약사에 보여주는 것이 기술이나 정보 유출이라는 인식이 있다”며 “이런 것은 빨리 잊어야 한다. 빅파마에게 평가받고 될성부른 것을 빠르게 골라낸 다음, 일부 연구 성과를 내서 기술수출로 이어지도록 해야 한다”고 말했다. 이어 “홀로 개발하는 시대가 아니다. 글로벌 기업의 전문가와 공유하고 거기서 상생할 길을 재설정하는 작업을 거쳐야만 상업화에 속도를 낼 수 있을 것”이라고 덧붙였다.

- 젬백스앤카엘, 알츠하이머병 치료제 글로벌 2상 환자 모집 완료
- [이데일리 송영두 기자] 젬백스(082270)앤카엘은 알츠하이머병 치료제 GV1001의 글로벌 2상 임상시험 환자 모집을 완료했다고 12일 밝혔다.이번 임상은 미국 83명, 유럽 116명으로 총 199명의 환자 등록을 완료했다. 경증 및 중등증 알츠하이머병 환자를 대상으로 GV1001 0.56mg 또는 1.12mg을 53주(12개월) 동안 피하 주사해 GV1001의 유효성 및 안전성을 평가한다. 1차 유효성 지표는 알츠하이머병 평가척도(ADAS-cog)다.젬백스는 미국 식품의약국(FDA)으로부터 임상시험계획서(IND)를 승인받아 스페인, 네덜란드, 폴란드 등 유럽 7개국으로 임상시험을 확대해 미국과 유럽의 총 43개 기관에서 2상 임상시험을 진행하고 있다.알츠하이머병은 치매를 일으키는 가장 흔한 신경퇴행성질환으로 신경세포 및 아교세포의 상호 커뮤니케이션 손상, 염증반응, 뇌세포 사멸 등을 일으켜 신경세포의 기능 손상과 감소가 나타나 결국 모든 일상생활 기능을 상실하게 된다. 최근 알츠하이머병의 진행 속도를 늦추며 인지능력 개선과 증상을 완화하는 약물이 시판됐으나 부작용 발생 위험도가 존재하는 등 여전히 미충족 수요가 높은 질환이다.젬백스의 GV1001은 현재까지 부작용과 이상 반응이 나타나지 않은 약물로 신경세포에서 베타 아밀로이드의 침착과 타우 단백질의 응축을 억제하는 등 알츠하이머병 치료에 대한 다양한 작용 기전을 가지고 있음을 확인한 바 있다. 최근 연구에서는 성선자극호르몬 방출호르몬 수용체(GnRHR)에 결합해 미세아교세포(microglia)와 성상교세포(astrocyte)를 직접적으로 조절하는 기전을 입증했다.젬백스 관계자는 “미국을 비롯해 총 8개국에서 글로벌 2상 임상시험을 시작하는 데 어려움도 있었지만, 환자 모집까지 완료하며 순조롭게 임상을 진행 중”이라며 “안전하고 근본적인 치료가 가능한 알츠하이머병 치료제 개발을 위해 모든 환자의 투약이 완료되고 최종 결과가 나올 때까지 최선을 다하겠다”고 말했다.한편, 젬백스는 또 다른 신경퇴행성질환이면서 희귀질환인 진행성핵상마비 (Progressive supranuclear palsy) 치료제 개발도 진행 중이다. 국내 최초 임상이기도 한 2상 임상시험의 환자 모집을 완료하여 연내 모든 환자의 투약이 마무리될 예정이다. 아울러 미국 식품의약국(FDA)으로부터 PSP 2상 임상시험계획(IND)을 승인받아 글로벌 신약 개발을 본격화했으며, 영국, 유럽 등에서도 임상시험 신청을 준비하고 있다.

- 아벨로스테라퓨틱스, 합성치사 항암제 전임상 발표...‘하반기 본임상 목표’
- [이데일리 유진희 기자] 혁신신약개발업체 아벨로스테라퓨틱스가 신개념 항암 치료제 본임상을 앞두고 막바지 작업에 들어갔다. 100억원 규모 이상의 시리즈B 유치와 전임상 결과 발표를 시작으로 하반기 본임상에 착수한다는 방침이다. (사진=아벨로스테라퓨틱스)◇시리즈B 막바지 조율...100억원 이상 유치 기대8일 업계에 따르면 아벨로스테라퓨틱스는 국내 유력 중견 투자사들이 참여한 시리즈B 단계 투자유치를 마무리 하기 위해 막바지 조율을 하고 있다. 100억원 이상 유치가 기대된다. 이와 함께 5일부터 10일(현지 시각)까지 엿새간 미국 샌디에이고에서 열린 ‘미국암연구학회(AACR) 2024’에 참가해 주요 파이프라인(신약 후보물질)의 비임상 연구 결과를 발표한다. 2021년 설립된 아벨로스테라퓨틱스는 합성치사와 DNA 손상복구, 세포주기를 조절하는 데 중점한 항암치료제 개발을 전문으로 하는 바이오벤처다. 합성치사는 두 개의 유전자가 동시에 변이, 억제, 손실이 일어나는 경우 세포 사멸이 일어나는 현상을 뜻한다. 기존 항암제 타깃이 되기 어려웠던 암 억제 유전자 돌연변이 등에 적용할 수 있어 GSK 등 글로벌 제약·바이오들이 주목하고 있다. 실제 최근 시리즈C에서만 1억 달러(약 1350억원)를 유치한 나스닥 상장사 바운드리스 바이오도 합성치사에 바탕한 항암치료제를 개발하고 있다. 지난 8일 나스닥 종가기준 시가총액은 약 3750억원이다. 이제 막 임상 1/2상 진행하는 바이오벤처로는 높은 평가다. 그만큼 합성치사 혁신신약에 대한 기대가 높다는 의미다. 아벨로스테라퓨틱스는 이 같은 기술에 기반한 항암치료제 후보물질 4개(AVS1001, AVS1002, AVS1003, AVS1004)를 확보하고 있다. AVS1001은 영국 MDI와 1년간 공동연구 이후 아벨로스테라퓨틱스가 자체연구를 통해 후보물질로 선정했다. 나머지 3개 파이프라인은 아벨로스테라퓨틱스의 독립적 연구를 통해 개발을 진행하고 있다. AACR 2024에서는 AVS1001을 포스터 공개한다. 항암작용을 유도하는 혁신신약으로서 단독 및 병용 요법에 대한 우수한 동물 약효 시험 결과를 발표한다.박영환 아벨로스테라퓨틱스 대표(오른쪽)와 오용호 디파이브테라퓨틱스 대표가 합성치사 항암제 개발을 위한 공동 연구계약을 체결한 뒤 기념촬영을 하고 있다. (사진=아벨로스테라퓨틱스)◇주축 박영환·최순규 공동대표, 회사 신뢰도 크게 높여 아벨로스테라퓨틱스의 역사는 짧지만, 박영환·최순규 공동대표가 회사의 신뢰도를 높이고 있다. 박 공동대표는 미국 머크 중앙연구소 감염질환 연구팀 리더, 대웅제약(069620) 연구본부장, 항암신약개발사업단 사업개발본부장, 국가항암신약개발사업단 사업단장 등을 역임했다. 한국 항암치료제 개발 분야의 최고 전문가 중 한 명으로 꼽힌다. 최 공동대표는 미국 바이엘 신약연구소와 PTC테라퓨틱스, GC녹십자 종합연구소 등을 거쳐 유한양행(000100) 중앙연구소장과 하나제약(293480) 연구본부장을 지냈다. 신약발굴에서 임상까지 전 과정에서 풍부한 경험을 쌓아온 인물이다. 이들은 바이오벤처의 ‘꽃’이라고 할 수 있는 기술수출과 미국 식품의약국(FDA) 허가도 다수 이끌었다. 박 대표는 머크 재직 당시 항생제와 희귀질환 치료제의 FDA 허가 등에 기여했다. 최 대표도 PTC 테라퓨틱스에서 개발한 희귀질환 SMA 치료제가 글로벌 제약사 로슈로 기술수출되고 신약으로 승인받는 데 일조했다. 이밖에도 유한양행 등에서 일하며, 글로벌 기술수출 등 다수의 성과를 이뤄냈다. 주요 투자사들이 아벨로스테라퓨틱스에 대한 투자를 아끼지 않는 배경이다. 아벨로스테라퓨틱스의 시리즈A 참여 투자기관은 △미래에셋캐피탈 △미래에셋벤처투자 △타임폴리오자산운용 △UTC인베스트먼트, △쿼드자산운 용등이며 약 100억원 규모를 유치했다. 최근 진행하는 시리즈B는 시리즈A 이상의 투자유치를 할 수 있을 것으로 관측된다. 두 공동대표는 “합성치사 분야라는 블루오션에서 차별화된 경쟁력으로 최고가 될 것”이라며 “이를 통해 암 환자들에게 더 나은 선택권을 제시해 그들의 삶의 질을 높이는 데 기여할 것”이라고 말했다.

- 혈액 속 살아있는 암세포 포획하는 세상 유일한 기업[싸이토젠 대해부①]
- [이데일리 석지헌 기자] 침이나 피 등 체액으로 암을 진단하는 액체생검 분야는 미국 매사추세츠공대(MIT)와 세계경제포럼(WEF)이 각각 선정한 ‘10대 유망 기술’이다. 액체생검 방식 중에서도 혈액 속을 돌아다니는 암세포(CTC)를 살아있는 채로 포획하는 기술은 그 동안 바이오 업계에선 불가능한 영역으로 여겨져 왔다. 미국의 바이오 기업 ‘셀서치’가 2012년 미국 식품의약국(FDA)으로부터 자성으로 암세포를 끌어모은 뒤 CTC를 수집하는 방법으로 가장 먼저 허가를 받았다. 하지만 자성으로 끌어들이다보니 세포의 변형이 일어났다. 이 기술을 사왔던 존슨앤드존슨도 3년 만에 포기했다. 이 문제를 해결한 게 싸이토젠(217330)의 창업자 전병희 대표다. 전병희 싸이토젠 대표.(제공= 싸이토젠)전 대표는 공학도 출신이다. 서울대학교 대학원에서 기계설계학으로 석사와 박사학위를 취득 후 인덕대학교 컴퓨터 응용 기계계열 교수를 역임했다. 바이오를 접한 건 삼성전기 고문직을 맡으면서다. 2007~2010년 삼성전기 바이오·전자장치 부문 고문을 맡으며 바이오공학에 뜻을 뒀다. 전 대표는 당시 난제였던 CTC 분리 기술에 대한 해법을 반도체에서 찾을 수 있을 것으로 확신, 2010년 싸이토젠을 창업했다. 지름 5㎛로 미세한 구멍을 뚫은 반도체 칩에 혈액을 통과시켜 암세포를 거르는 방식을 고안한 것이다. 순전히 공대 출신 관점에서 암세포를 제대로 잡기만 하면 된다는 생각으로 개발에 몰두했다는 설명이다. 일반적인 암세포의 크기는 7㎛ 안팎이다. 7㎛ 안팎인 암세포는 걸러지고, 이보다 작은 적혈구와 백혈구는 빠져나간다. 세포가 구멍 가장자리에 긁혀 손상되지 않도록 특수 코팅 처리도 했다. 이후 싸이토젠은 암세포를 추출하는 기계, 세포를 염색하는 기계, 이 세포들을 분석하는 기계를 독자적으로 개발했다. 모든 과정은 자동이다. 혈액 속 암세포를 살아있는 상태로 채집하는 건 파괴된 암세포가 남긴 유전자 정보(DNA)보다 얻을 수 있는 정보가 훨씬 많다. 순도가 높은 데다 원발암(최초 발생암) 정보도 확인할 수 있다. 암 환자 90% 이상이 암 전이로 사망한다는 점을 고려하면 싸이토젠 기술이 갖는 의미가 상당하다고 볼 수 있다. 전 대표는 “직경 10㎜에 미세 구멍이 60만개가 균일하게 뚫려있다. 별다른 압력 없이 여기에 혈액을 떨어뜨리면 CTC를 손상없이 잡아낼 수 있다”며 “액체생검을 이용해 DNA 레벨에서 유전체 검사를 제공하는 회사는 많지만, 살아있는 세포에서 DNA와 RNA, 프로틴을 모두 추출해 유전체 정밀검사를 할 수 있는 곳은 사실상 싸이토젠이 유일하다”고 설명했다.전 대표는 현재 32명의 연구개발 인력과 함께 하고 있으며 이 중에는 기계공학 전공자도 2명이 포함돼 있다. 반도체 장비 업체에서 15년 간 근무한 경력자도 있다. 최근 최대주주가 캔디엑스홀딩스로 바뀌면서 공동대표 체제가 됐다. 캔디엑스홀딩스에는 엑세스바이오(950130)와 메리츠증권(008560), 홍콩계 PE인 엑셀시아캐피탈코리아 등이 주축으로 참여했다. 기존 사업은 전 대표가, 신규 사업과 경영기획본부는 사철기 대표가 각각 맡는다. 사 대표는 유한양행에서 R&D와 관련된 여러 영역에서 책임자로 일해왔으며, 유한메디카의 대표이사, 비씨월드제약의 상임고문을 역임했다.
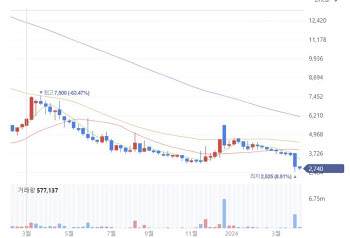
- 조기 기술수출 선언한 카이노스메드..."파킨슨 신약 美·日·中 기업과 논의 중"
- [이데일리 송영두 기자] 카이노스메드는 개발 중인 파킨슨병 치료제의 조기 기술수출을 예고했다. 이르면 상반기 기술수출 승부수를 던질 전망이다. 하지만 기술수출은 일차적으로 임상 데이터가 우수해야 하는 만큼 현재 진행 중인 미국 임상 2상 결과가 관건이 될 것이라는 분석이다.9일 제약바이오 업계에 따르면 카이노스메드(284620)는 파킨슨병 치료제로 개발 중인 ‘KM-819’ 조기 기술이전 추진을 선언했다. 최근 주가 하락에 따른 주주들의 불만이 높아지자, 회사 중요 사업 진행 현황을 발표한 것이다.카이노스메드는 지난해 4월 5일 6850원의 주가를 기록했지만, 올해 들어 4000원대로 떨어진 뒤 별다른 반등없이 지난 4일 2740원대까지 떨어졌다. 2014년 중국 기술수출됐던 자체개발 에이즈 치료제 KM-023이 지난해 중국 의료보험시장을 뚫으면서 호재가 발생했고, 중국 장수아이디가 글로벌 판권까지 도입하면서 큰 기대를 모았다. 중국 에이즈 치료제 시장 규모는 2023년 73억 위안(약 1조3000억원)에서 2027년 112억 위원(약 2조원)으로 성장할 것으로 예상된다.하지만 이런 호재에도 카이노스메드 주가는 크게 반등하지 못했다. 회사가 중국에서 에이즈 치료제 판매로 받을 수 있는 규모가 매출액의 2%에 불과해 실질적인 매출 상승에 영향을 주지 못했기 때문이다. 실제로 KM-023은 지난해 중국 시장에서 전년대비 2배가 넘는 130억원 이상의 판매를 기록한 것으로 알려졌는데, 2023년 카이노스메드 매출액은 약 2억6700억원에 불과했다. 매출은 모두 에이즈 치료제 로열티였다. 치료제를 개발하고 판매가 이뤄지고 있음에도 주가와 연결이 되지 않은 이유다.카이노스메드 주가 추이.(자료=네이버페이증권)◇반등 카드는 ‘파킨슨 치료제’...상반기 기술이전 추진카이노스메드는 파킨슨병 치료제 KM-819의 조기 기술이전을 추진 중인 사실을 공개했다. 주가 반등과 주주들의 불만을 가라앉히기 위한 수단으로 기술이전을 언급한 모양새다. 회사 측은 임상 2상 중간 결과가 나오면 그 데이터를 바탕으로 기술이전에 나선다는 계획이다. 카이노스메드 관계자는 “파킨슨병 치료제로 개발하고 있는 KM-819는 현재 미국에서 임상 2상을 진행하고 있다”며 “임상 2상은 Part 1a, Part 1b, Part 2로 분류된다”고 설명했다.회사 측에 따르면 Part 1a는 건강한 노인층 18명을 대상으로 KM-819의 400mg, 600mg, 800mg 3가지 용량에 대한 안전성을 확인하는 단계다. 800mg 용량까지 안전함을 모두 확인했다. Part 1b는 파킨슨병 환자 대상으로 실시한 용량결정 시험단계다. 200mg, 300mg 용량으로 8명 환자에게 투여를 전부 완료한 상태다.Part 1b 데이터 분석이 끝나고 미국 식품의약국(FDA)에 part2를 위한 임상계획서를 제출하면 7월 이후에 part 2가 개시될 것으로 예상된다. 회사 관계자는 “Part 1b의 데이터 분석 결과는 올해 상반기 내로 나올 것으로 예상한다. 이 결과를 바탕으로 당사는 조기 기술이전을 추진하고자 한다”고 말했다.◇임상 2상 유의미 결과 기대...美·日·中 기업과 논의 중회사 측은 임상 2상 중간 결과에 기대를 보이는 모습이다. 카이노스메드 관계자는 “KM-819에 대한 작용기전, 전임상 데이터, 임상 1상 데이터, 그리고 올해 상반기 내 분석될 임상 2상 결과들을 바탕으로 글로벌 제약사 등과 라이센스 아웃을 논의 중”이라고 말했다.특히 “전임상 데이터에서는 파킨슨병 동물모델에서 KM-819 약물 치료 효과를 충분히 확인했고, 임상 1상 데이터 및 임상 2상 Part 1a를 통해 건강한 사람에게서 약물 안전성도 고용량까지 확인했다”며 “이번 Part 1b를 통해 환자에서 확인되는 유효성과 안전성, 최적의 용량이 결정되면 임상적으로 유의미한 결과를 확인할 수 있을 것으로 기대한다”고 강조했다. 업계 관계자는 “파킨슨 치료제는 아직 상용화된 제품이 없고 개발 성공 확률이 낮은 만큼 기회는 열려있다”면서도 “상반기에 나올 임상 2상 중간 데이터가 굉장히 좋은 수준이라면 분명 기술이전에 적극적인 기업들이 있을 것이다. 하지만 데이터 발표 후에도 기술이전에 속도가 나지 않는다면 카이노스메드 입장에서 치료제 개발을 지속할 이유가 없어질 것”이라고 설명했다.현재 카이노스메드는 미국, 일본, 중국 등 각 지역 다수 제약사 및 바이오텍들과 기술이전 논의를 진행 중이라고 밝혔다. 다만 회사 측은 연내 기술이전이 가능하다고 판단하는 이유에 대해서는 “답변하기 조심스럽다”면서 구체적인 언급을 하지 않았다. 또 지난해 감사보고서에서 적정 판정을 받았지만, 계속기업 존속불확실성이 기재된 만큼 이를 타개할 전략에 대한 질문에 대해서도 “(계속기업 존속불확실성) 해소를 위해 다양한 시나리오를 가동 중이며, 아직 확정된 사항이 아닌 만큼 상세한 내용을 언급하긴 어렵다”면서 “연내 기술이전 및 해외자본 유치에 저력을 다할 것”이라고 말했다.
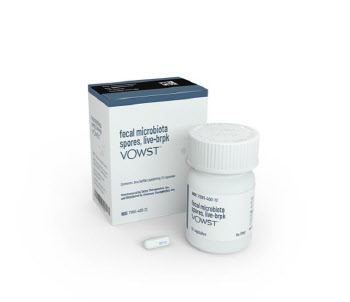
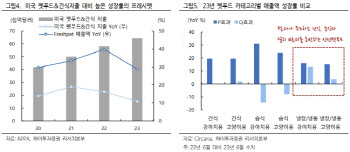
- 2호 마이크로바이옴 신약, 대박조짐…국내업계에 훈풍 예고
- [이데일리 나은경 기자]글로벌 2호 마이크로바이옴 치료제의 매출이 빠르게 성장하면서 한동안 주춤했던 마이크로바이옴 업계에 대한 기대감도 부풀고 있다. 특히 올해는 글로벌하게 3·4·5호 마이크로바이옴 신약이 의약당국의 승인을 받을 것으로 예상돼 국내 마이크로바이옴 업계에도 훈풍이 불어닥칠 전망이다.◇보우스트, 마이크로바이옴 반등 신호탄 쐈다4일 세레스 테라퓨틱스(이하 ‘세레스’)에 따르면 지난해 ‘보우스트’(VOWST)의 매출은 1960만 달러(약 264억원)를 기록했다. 지난해 6월 출시, 6개월만에 2000만 달러에 가까운 매출을 낸 셈이다. 설사병의 일종인 재발성 클로스트리디움 디피실 감염증(CDI) 치료제 보우스트는 CDI에 대한 항균 치료 후 재발 방지 용도로 승인받아 1차 치료제도 아니어서 더 눈길을 끈다.미국 바이오텍 세레스 테라퓨틱스의 CDI 치료제 ‘보우스트’ (사진=세레스 테라퓨틱스)바이오 업계 관계자는 “마이크로바이옴이 새로운 모달리티(치료접근법)이고 이제 막 출시된 상황이라는 점을 감안하면 보우스트의 지난해 매출은 향후의 성장세를 기대해 볼만한 규모”라고 말했다.보우스트보다 먼저 세계 최초 마이크로바이옴 치료제로 FDA의 허가를 받은 것은 페링 테라퓨틱스의 ‘리바이오타’(REBYOTA)다. 리바이오타는 건강한 사람의 대변을 제조해 그 미생물총을 내시경을 통해 환자의 장내에 뿌리는 방식의 CDI치료제다. 이와 달리 ‘2호 마이크로바이옴 치료제’인 보우스트는 첫 경구용 마이크로바이옴 치료제여서 편의성이 높다. 감염 및 알레르기 리스크가 있는 분변이식 대비 안전성도 높다는 장점도 있다.이 같은 이유로 마이크로바이옴 업계에서는 보우스트를 사실상의 첫 마이크로바이옴 치료제로 여기고 첫 해 매출 규모에 주목해왔다. 특히 올해는 보우스트의 성장세가 더 커질 것으로 기대되는 상황이다. 세레스는 네슬레그룹의 헬스케어·바이오 전문기업인 네슬레 헬스사이언스와 공동으로 보우스트의 상업화를 진행 중인데, 올해 더 공격적인 의사 교육 및 마케팅을 진행하겠다는 전략이다.허가를 앞둔 차기 마이크로바이옴 치료제들의 임상 데이터도 고무적이다. 스페인 바이오텍 마이크로바이오믹(Mikrobiomik)이 개발 중인 CDI치료제 ‘MBK-01’은 임상 3상 중간분석 결과 환자 모집이 완료되기 전임에도 강력한 효능을 보이면서 조기 임상 완료 가능성까지 제기됐다. 지금은 유럽의약품청(EMA)에 허가를 받기 위한 준비를 진행 중이다. 중국 오사이바이오파마(OSAI Biopharma)의 세균성질염 치료제 ‘Lc262-1’ 후보물질도 임상 3상에서 긍정적 결과를 입증, 중국 국가약품감독관리총국(NMPA)의 시판 승인 절차를 밟고 있다.◇부진했던 韓업체도 ‘글로벌 훈풍’ 수혜 기대지난해 글로벌 마이크로바이옴 바이오텍들의 폐업과 매각이 잇따랐고, 한국 마이크로바이옴 업계도 부진을 면치 못했다. 실제 마이크로바이옴 대장주인 고바이오랩, CJ바이오사이언스, 지놈앤컴퍼니의 주가는 지난해 평균 30.8% 하락했다. 반면 같은 기간 KRX헬스케어 지수는 오히려 20.1% 상승했다.(그래픽=이데일리 이미나 기자)임상 중단, 기술이전 지연 등으로 대장주로 꼽히는 마이크로바이옴 업체들의 사업화가 지연된 것이 가장 큰 원인으로 분석된다. 고바이오랩(348150)은 지난해 6월 주력 파이프라인 중 하나로 미국 FDA의 허가를 받아 임상 2a상을 진행 중이던 궤양성대장염 치료제 후보물질 ‘KBLP-007’의 개발을 중단한다고 밝혔다. 지놈앤컴퍼니(314130)는 상장 당시 2023년 기술이전을 통한 흑자전환을 목표로 했지만 임상 진행이 늦어지면서 여전히 500억원대 적자를 내고 있다. 상장 이전인 2019년 이뤄진 LG화학(051910)과의 기술이전 계약 외엔 아직까지 눈에 띄는 기술이전 성과가 없는 상태다. CJ 바이오사이언스(311690)(옛 천랩)의 경우 지난해 10월 처음 임상 1상 첫 환자 투약을 했을 정도로 경쟁사에 비해 연구·개발 진도가 더딘 편이다. 하지만 최근 글로벌 마이크로바이옴 시장에 긍정적인 소식들이 쌓이고 있어 국내 기업들도 촉각을 곤두세우는 분위기다.지난해 글로벌 마이크로바이옴 시장에서는 글로벌 빅파마들의 인수·합병(M&A) 및 파트너 찾기가 그 어느 때보다 활발했다. 발생한 M&A 건수가 과거 연간 평균값의 3배로 늘어났을 정도다. 특히 독일 제약사 베링거인겔하임은 T3파마슈티컬스를 약 5억 달러(약 7000억원)에 인수했는데, 이는 이제까지 마이크로바이옴 시장에서 이뤄진 가장 큰 규모의 M&A다. 덴마크의 노보 노디스크도 지난해 8월 스나이퍼바이옴(SNIPR BIOME)과 신규 유전자치료제 개발을 위한 계약을 확장한 바 있다. 이 같은 분위기가 지속된다면 그간 연기됐던 국내 마이크로바이옴 업계의 기술수출도 연내는 기대해 볼 수 있다는 분석이다.시장 반등의 분위기를 타고 비상장 마이크로바이옴 바이오텍들도 하나 둘씩 기업공개(IPO)를 준비 중이다. 셀트리온(068270)과 신약개발 공동연구 중인 리스큐어바이오사이언시스가 내년 코스닥 상장을 목표로 상반기 중 기술성 평가를 신청할 예정이고, 유한양행(000100)이 59.6%의 지분을 가진 자회사 에이투젠도 코스닥 상장을 준비하고 있다.

- 나이벡, ‘재생기전 염증성 장질환 치료제’ 연구개발 성과 발표
- [이데일리 박순엽 기자] 나이벡은 ‘IBD INNOVATE Conference 2024(염증성 장질환 콘퍼런스)’에 참가해 자체 개발 중인 재생기전 염증성 장질환 치료제 ‘NP-201’에 대한 연구개발 성과를 발표한다고 9일 밝혔다. NP-201의 미국 임상을 추진 중인 나이벡은 이번 콘퍼런스를 통해 임상기관 선정을 위한 네트워킹도 병행할 계획이다. 나이벡 CI (사진=나이벡)염증성 장질환 콘퍼런스는 미국의 ‘염증성 장질환협회(Crohn‘s&Colitis Foundation)’ 주최로 매년 개최되는 글로벌 학술대회다. 전 세계에서 염증성 장질환 치료제를 연구하는 석학들과 글로벌 제약사, 전문 연구기관들이 대거 참가해 나이벡은 NP-201에 대한 FDA 임상을 수행할 임상기관 후보군을 압축할 수 있을 것으로 기대하고 있다. 이번 콘퍼런스에서 나이벡은 펩타이드 기반 재생기전의 염증성 장질환치료제 NP-201에 대한 임상 및 전임상 시험 데이터를 공개할 예정이다. NP-201은 지난해 폐섬유증 치료제를 적응증으로 글로벌 임상1상을 진행하며 이미 인체 안전성이 입증됐다. 글로벌 임상에선 총 32명의 건강한 성인을 대상으로 피하주사(SC) 제형의 NP-201과 위약을 병용 투여했다. 투약 용량은 환자군 별로 100mg, 200 mg, 300 mg, 400 mg 투여가 진행됐다. 시험결과 NP-201은 고농도에서 흡수, 분포, 배설까지 전혀 이상이 없었으며, 반감기 또한 적정시간 유지되는 것으로 나타났다.전임상 시험에서도 NP-201은 효능이 확인됐다. 만성동물 모델에서 용량의존적으로 장 길이 증가, 질병활성도 감소, 점막재생 바이오마커의 발현이 증가했다. 이는 NP-201이 염증 억제 효능뿐 아니라 손상된 세포조직을 재생해 근본적인 치료도 가능케 한다는 게 회사 측 설명이다.나이벡 관계자는 “이번 콘퍼런스 기간 최대한 많은 미국 내 염증성 장질환 전문기관들과 접촉할 예정이며, 이중 NP-201의 FDA 임상에 최적화된 임상기관을 선정할 계획”이라며 “NP-201은 기존에 치료제와는 차별화된 재생기전을 보유하고 있어 많은 임상수행 기관들이 관심을 표명할 것으로 예상된다”고 말했다.염증성 장질환은 대장에 염증 및 궤양이 생기는 난치성 질환이다. 인체에 침입한 균을 공격하는 백혈구가 대장이나 다른 소화기관을 적으로 간주하고 공격해 발생하는 자가면역 질환 중의 하나다. 업계에 따르면 궤양성 대장염과 크론병을 포함한 염증성 장질환 치료제의 글로벌 시장규모는 약 40조원으로 추정된다.